STAT115 Chapter 2.1 Protein Wave
Summary
TLDRThis course introduction to computational biology and bioinformatics at Harvard explores the history and evolution of bioinformatics, from Fred Sanger’s pioneering work on protein sequencing in 1955 to the rise of AI-driven tools like DeepMind’s AlphaFold. The lecture covers key developments in protein sequence alignment, the establishment of the Protein Data Bank, and the challenges of predicting protein structures. It highlights the significance of computational algorithms in understanding protein function and structure, with a focus on how AI is transforming the field and solving previously unsolvable problems in protein structure prediction.
Takeaways
- 😀 Bioinformatics is a highly useful skill for statisticians, computer scientists, and biologists, providing valuable tools for research and analysis.
- 😀 The history of bioinformatics started with protein sequence analysis, with Fred Zenger's 1955 sequencing of bovine insulin paving the way for drug synthesis.
- 😀 Pairwise sequence alignment algorithms like Needleman-Wunsch were developed to compare protein sequences and measure their similarity.
- 😀 In 1973, the Protein Data Bank (PDB) was established to store solved protein structures, enabling correlation between protein sequences, structures, and functions.
- 😀 As more protein sequences became available, database search tools like BLAST were created to enable fast searches and avoid the computational cost of pairwise alignment.
- 😀 The Critical Assessment of Structure Prediction (CASP) competition, established in 1994, enabled researchers to test and improve computational methods for protein structure prediction.
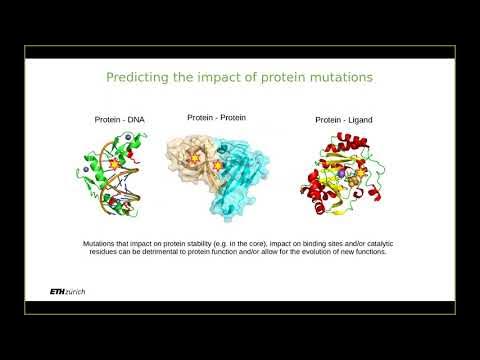
- 😀 Functional domains within proteins help in predicting structures, as similar domains often suggest similar functions and structures.
- 😀 By 2012, the state of the art in protein structure prediction showed that methods like HHpred and Rosetta could predict easier protein structures with high accuracy.
- 😀 The introduction of AlphaFold in 2018 revolutionized protein structure prediction, leveraging AI to predict protein structures with much greater accuracy.
- 😀 By 2020, AlphaFold 2 achieved near-experimental accuracy in protein structure prediction, pushing the field closer to solving the problem of structure prediction for single proteins.
- 😀 Challenges remain in predicting protein-protein interactions and the structures of large molecular complexes, but advancements in AI and computational biology continue to drive improvements.
Q & A
What is the primary focus of the Bioinformatics course mentioned in the transcript?
-The primary focus of the course is to provide students with an introduction to computational biology and bioinformatics, covering key topics such as protein sequence alignment, structure prediction, and the application of computational tools like databases and AI in bioinformatics.
Why is bioinformatics considered an essential skill for biologists and those in computational fields?
-Bioinformatics is essential because it enables biologists to analyze and interpret large datasets of biological information, especially protein sequences and structures. It also equips those in computational fields to develop tools and algorithms that support biological research, making it a crucial skill for many disciplines.
How did Fred Sanger contribute to the development of bioinformatics in 1955?
-Fred Sanger developed the technology to sequence proteins, starting with the sequencing of bovine insulin. His work enabled the synthesis of insulin as a drug and laid the foundation for the study of protein sequences, a key component of bioinformatics.
What is pairwise sequence alignment and why is it important in bioinformatics?
-Pairwise sequence alignment is a method used to compare two protein or DNA sequences to identify similarities or differences. It is important because it helps determine whether new sequences have been encountered before and can provide insights into the function and structure of the proteins involved.
What role does the Protein Data Bank (PDB) play in bioinformatics?
-The Protein Data Bank (PDB) stores experimentally solved 3D structures of biological macromolecules, including proteins. Researchers can access the PDB to find known structures and use them as references for predicting the structures of new proteins.
What was the purpose of developing algorithms like BLAST in bioinformatics?
-BLAST (Basic Local Alignment Search Tool) was developed to enable fast searches in large sequence databases. Instead of comparing sequences pairwise, BLAST allows researchers to quickly identify sequences that are similar to a given query, making the search for related proteins much more efficient.
What is the CASP competition, and how does it contribute to protein structure prediction?
-The CASP (Critical Assessment of Structure Prediction) competition is held every two years to evaluate the accuracy of protein structure prediction algorithms. Participants are asked to predict the 3D structures of proteins based on their sequences, and their predictions are tested against experimentally determined structures, helping to advance the field.
What major breakthrough did AlphaFold achieve in protein structure prediction?
-AlphaFold, developed by DeepMind, achieved a major breakthrough by predicting protein structures with remarkable accuracy using AI. In the 2020 CASP competition, AlphaFold's predictions were shown to have a Global Distance Test score above 90, which is considered equivalent to experimentally determined structures.
Why are protein-protein interactions still considered a challenging problem in bioinformatics?
-Protein-protein interactions involve the complex behavior of multiple proteins interacting in a biological system. Predicting how these interactions occur, especially in large complexes, is still a challenge because the sheer complexity of these interactions requires understanding both the individual proteins and how they form functional groups together.
How does AI contribute to advancing the field of bioinformatics, particularly in protein structure prediction?
-AI, particularly through tools like AlphaFold, has advanced bioinformatics by learning from large datasets of protein sequences and structures. Unlike traditional methods, AI-based approaches can automatically learn the rules of protein folding and predict structures with high accuracy, reducing the need for manual input from biochemists and accelerating research.
Outlines

This section is available to paid users only. Please upgrade to access this part.
Upgrade NowMindmap

This section is available to paid users only. Please upgrade to access this part.
Upgrade NowKeywords

This section is available to paid users only. Please upgrade to access this part.
Upgrade NowHighlights

This section is available to paid users only. Please upgrade to access this part.
Upgrade NowTranscripts

This section is available to paid users only. Please upgrade to access this part.
Upgrade Now




5.0 / 5 (0 votes)